
The integration of artificial intelligence with personal protective equipment creates a complex regulatory landscape, particularly for the European market. CE marking for AI-integrated smart masks is not merely a compliance exercise but a rigorous process that validates safety, performance, and data integrity. For manufacturers bringing these advanced products to the EU/EEA market, understanding the multi-faceted certification pathway is crucial to avoid costly delays and ensure market access.
CE marking for AI-integrated smart masks requires compliance with multiple European regulations simultaneously: the Medical Devices Regulation (MDR) 2017/745 for health monitoring features, the Personal Protective Equipment Regulation (EU) 2016/425 for respiratory protection, the Radio Equipment Directive (RED) 2014/53/EU for wireless connectivity, and data protection requirements under the General Data Protection Regulation (GDPR), creating a complex conformity assessment that demands careful planning and documentation. Successfully navigating this process involves classifying the device correctly, selecting the appropriate conformity assessment procedure, and working with notified bodies that have expertise across all relevant domains.
The European market for smart wearables is projected to reach €25 billion by 2025, with stringent regulations ensuring product safety. According to guidance from the European Commission, an AI-integrated mask that provides health data analysis (like respiratory rate alerts) is typically classified as a Class I or IIa medical device under MDR, while its filtration function falls under PPE Category III, representing one of the highest risk categories. Let's explore the systematic approach to achieving CE marking for these hybrid products.
What is the Correct Classification for Your Smart Mask?
The first and most critical step is determining the correct regulatory classification, as this dictates the entire conformity assessment pathway. Misclassification at this stage can lead to application rejections or, worse, market recalls.

How Do MDR Rules Apply to AI Health Features?
Under the Medical Devices Regulation (MDR), classification depends on the intended purpose. If your mask's AI provides information for "monitoring, prediction, prognosis, or alleviation of disease" (e.g., detecting abnormal breathing patterns suggestive of respiratory distress), it is a medical device. Key rules include:
- Rule 10: Active therapeutic devices and devices for diagnosis or monitoring. Devices intended to "monitor vital physiological processes" are generally Class IIa, unless monitoring changes that could lead to immediate danger, which could be Class IIb.
- Rule 11: Software intended to provide information for diagnostic or therapeutic decisions. Standalone AI software is typically Class IIa or higher.
A formal "Intended Purpose" statement is required to anchor this classification. According to the Medical Device Coordination Group (MDCG) guidance, claims like "detects signs of respiratory fatigue" or "monitors breathing rate for health insights" will trigger medical device classification.
What PPE Category Applies to the Filtration Function?
Concurrently, the mask's primary protective function as respiratory protective equipment (RPE) is regulated under the PPE Regulation (EU) 2016/425. Filtering facepieces (FFP1, FFP2, FFP3) designed to protect against hazardous aerosols are classified as Category III. This is the highest risk category because it involves risks that may cause very serious consequences such as death or irreversible damage to health. This classification mandates the involvement of a Notified Body for the product's type-examination and ongoing surveillance of production.
Which Conformity Assessment Procedure Must Be Followed?
For a Category III PPE with Class IIa medical device features, a hybrid conformity assessment procedure is required, combining modules from both regulatory frameworks.
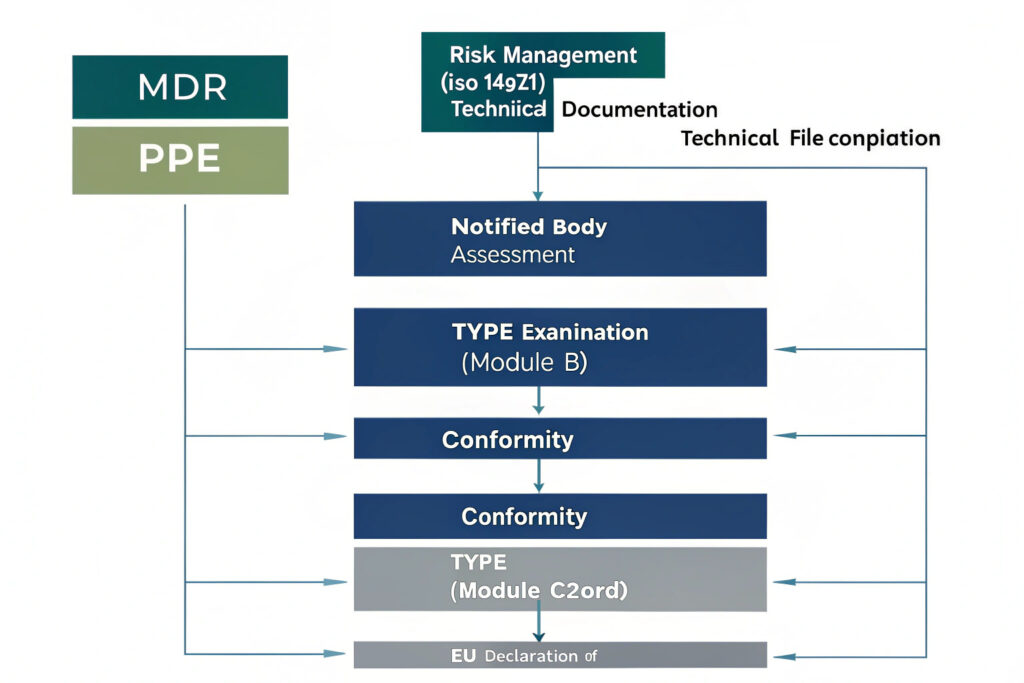
What is the Typical Module Combination for Such a Product?
The most common route involves:
- Module B: EU Type Examination (for PPE Regulation): A Notified Body examines the technical design of the mask's protective aspects and verifies it meets the essential health and safety requirements (EHSRs) of the PPE Regulation, resulting in an EU Type Examination Certificate.
- Annex IX Chapter I (for MDR - Class IIa): This involves a conformity assessment based on a quality management system (QMS) and assessment of the technical documentation for the medical device aspects. The same Notified Body can often perform both assessments if it holds appropriate designations.
- Module C2: Conformity to Type based on Internal Production Control plus Supervised Product Checks (for PPE): This ensures ongoing production matches the certified type, with periodic audits and product checks by the Notified Body.
This combined approach satisfies both regulations. Guidance from notified bodies like BSI or TÜV SÜD is invaluable in selecting the precise module sequence.
How is a Single Notified Body Selected for Both Assessments?
Few Notified Bodies are designated under both the MDR and the PPE Regulation for the relevant product codes. You must identify a body with the correct scope of designation (e.g., PPE Regulation Code 11.03 for filtering respiratory devices and MDR codes for active therapeutic/diagnostic devices). It is critical to conduct an "Application Readiness Review" with the chosen Notified Body early in the development process to align on the classification and assessment strategy.
What Technical Documentation is Required?
The technical documentation is the cornerstone of the CE marking process. For a smart mask, it becomes a multi-volume dossier integrating electrical, software, mechanical, and biological safety data.

What are the Critical Components of the Technical File?
The documentation must satisfy both MDR Annex II and the general requirements of the PPE Regulation. Essential sections include:
- Device Description & Specifications: Full details of all components, including AI algorithms, sensors, and connectivity.
- Risk Management File (per ISO 14971): A comprehensive analysis covering mechanical, electrical, biological, data security, and clinical risks. This must include specific risks of AI (e.g., algorithm bias, false positives/negatives).
- Software Documentation (per IEC 62304): Includes the Software Development Lifecycle (SDLC) plan, architecture, verification/validation testing, and for AI/ML, details on data provenance, training, and performance evaluation.
- Usability Engineering File (per IEC 62366-1): Demonstrates the device is safe and effective for use, considering the user interface, app, and instructions.
- Performance & Safety Test Reports: Filtration efficiency (EN 149), breathing resistance, flammability, biocompatibility (ISO 10993), electrical safety (IEC 60601-1), and EMC/Radio (per RED).
- Clinical Evaluation Report (for MDR): Scientific evidence validating the medical claims, which may include literature review, bench testing, and possibly a clinical investigation.
How is the AI/ML System Specifically Documented?
The AI component requires transparent documentation. The IMDRF's guidance on Software as a Medical Device (SaMD) is a key reference. You must document:
- Algorithm Change Protocol: How updates will be managed and validated.
- Data Management: Description of training, tuning, and test datasets, including demographics and measures to address bias.
- Performance Metrics: Detailed results for sensitivity, specificity, accuracy, and robustness across different populations and conditions.
- Clinical Validation Plan & Report: Evidence that the AI output is clinically meaningful and safe.
How Do Data Protection (GDPR) and Radio Equipment (RED) Requirements Integrate?
The smart mask's connectivity and data processing introduce two additional regulatory layers that must be woven into the conformity assessment.

What GDPR Compliance Must Be Demonstrated?
If the mask processes personal health data in the EU, GDPR applies. Compliance should be integrated into the technical documentation:
- Data Protection by Design and by Default: Describe technical measures (encryption, anonymization) and organizational measures in the risk management file.
- Legal Basis for Processing: Typically, explicit user consent will be required for processing health data. The user interface and instructions must facilitate valid consent.
- Data Processing Agreement (DPA): If using cloud services, a DPA with the provider is needed.
While Notified Bodies do not certify GDPR compliance, demonstrating a robust approach is essential for the overall safety and market acceptance of the device. Resources from the European Data Protection Board (EDPB) provide guidance.
What are the RED Requirements for Bluetooth/Wi-Fi Connectivity?
The Radio Equipment Directive (RED) ensures radio equipment doesn't cause harmful interference and protects user health (SAR - Specific Absorption Rate). Key steps include:
- Testing to Harmonized Standards: e.g., ETSI EN 300 328 for Wi-Fi, EN 300 440 for Bluetooth.
- Assessment of Safety & Health (Article 3.1a): Compliance with safety standards like IEC 62368-1 and SAR testing if applicable.
- Assessment of EMC (Article 3.1b): Electromagnetic Compatibility testing.
- Assessment of Efficient Use of Spectrum (Article 3.2): Radio testing.
Often, the Notified Body for the MDR/PPE can subcontract or coordinate RED testing, but it must be included in the overall conformity assessment.
What is the Process After Certification?
Achieving the CE mark is not the end. Significant post-market obligations are required to maintain compliance.

What Post-Market Surveillance (PMS) is Required?
Both MDR and PPE Regulation impose stringent PMS requirements:
- Proactive Collection: Systems to collect data on device performance in the field, user feedback, and literature reviews.
- Periodic Safety Update Report (PSUR): For Class IIa+ devices under MDR, an annual report summarizing PMS data and conclusions.
- Vigilance and Reporting: Serious incidents and field safety corrective actions must be reported to the relevant Competent Authorities (e.g., MHRA, BfArM) and the Notified Body within strict timelines.
The PMS plan, documented in the technical file, must be actively executed.
How are Software and AI Updates Managed?
Any update to the AI algorithm or software that affects the device's safety or performance requires a documented change control process. Significant updates may require re-submission to the Notified Body for review under the certified quality management system. The Algorithm Change Protocol established during certification will define the criteria for what constitutes a significant change.
Conclusion
Navigating CE marking for an AI-integrated smart mask is a complex but manageable process that demands an integrated, strategic approach from the outset. Success hinges on precise classification, selecting a capable Notified Body, building comprehensive technical documentation that satisfies MDR, PPE, RED, and GDPR requirements, and establishing robust post-market systems. Early engagement with regulatory experts and your chosen Notified Body is the most critical factor in efficiently steering this hybrid product to the European market.
Ready to chart the regulatory path for your AI-integrated smart mask? Contact our Business Director, Elaine, at elaine@fumaoclothing.com. Our regulatory affairs team has experience navigating the complex intersection of MDR, PPE, and digital health regulations and can guide you from concept to compliant market entry.


















